
In active pharmaceutical ingredient (API) development, solid form selection is one of the most influential factors in a molecule’s success. The chosen solid form has important implications for manufacturability, product behaviour, and intellectual property.
When the optimal solid form is not considered early on in an API development project, challenges with filtration, stability, batch variability, and more may surface as the project progresses, leading to avoidable delays and costly rework.
In this blog, we’ll take a closer look at why solid form selection matters in API development, then explore some of the screening and evaluation techniques that can be used to identify the most ideal solid form for a particular project.
A closer look at solid form selection
An active pharmaceutical ingredient (API) does not exist in just one physical state. The same molecule can adopt different packing arrangements, known as polymorphs, and can be further modified by salt, cocrystal, hydrate, or solvent formation.
Each of these solid form variations can dramatically alter the molecule’s physical and chemical properties, influencing critical factors like solubility, stability, and bioavailability. For instance, one version of the API may be more bioavailable than another.
As these traits directly affect formulation and commercial viability, understanding solid state variability early in development is essential.
The impact of internal structure: Amorphous vs. crystalline
The European Medicines Agency (EMA) classifies API by appearance (particle shape) and internal structure (molecular arrangement).
- Amorphous forms are the most disorganised. They can offer improved solubility, but potentially lower shelf life.
- Crystalline forms are more orderly and tend to be more stable, but certain polymorphs may have low melting points or processing limitations.
A multitude of analytical techniques may be used to characterise solid forms, including X-ray powder diffraction (XRPD), differential scanning calorimetry (DSC), thermogravimetric analysis (TGA), high-performance liquid chromatography (HPLC), optical microscopy, and hot stage microscopy (HSM), among others. The most advantageous form depends on specific project objectives, as selecting the right one often requires a balance between competing attributes.
Characteristics that define solid form performance
Solid APIs can be classified through five property characteristics:
- Thermodynamic, including melting temperatures, enthalpy, and entropy
- Packing, including molar volume and density and refractive indices
- Kinetic, including dissolution rate and stability
- Surface, including habit/size, hygroscopicity, and interfacial tensions
- Mechanical, including hardness and compactibility
A strong understanding of each of these parameters is crucial for guiding solid form development.
Biopharmaceutical considerations for early development
Solid form selection is also influenced by the Biopharmaceutics Classification System (BCS), which categorises APIs based on solubility and permeability. These classifications are summarised in the table below.
| BCS class | Solubility | Permeability | Absorption pattern | Examples |
|---|---|---|---|---|
| I | High | High | Well absorbed | Metoprolol, diltiazem, propranolol |
| II | Low | High | Well absorbed | Phenytoin, nifedipine, danazol |
| III | High | Low | Variable | Cimetidine, acyclovir, catopril |
| IV | Low | Low | Poorly absorbed | Hydrochlorothiazide, taxol, furosemide |
Many new chemical entities (NCEs) fall into classes II and IV, with low solubility. For class II APIs, any improvement to solubility, such as by salt or cocrystal formation, would be expected to improve efficacy. Class IV APIs are the most challenging, since absorption and solubility are both poor. Particle size reduction and other tactics would be beneficial in this case.
As so many factors go into solid form selection, and each solid form presents distinct advantages and disadvantages, a methodical approach to solid form screening and optimisation is crucial for selecting the optimal form and mitigating downstream challenges.
What goes into choosing the right solid form?
Solid form selection and optimisation may consist of five important elements: salt and cocrystal screening, pre-formulation evaluation, polymorphism investigations, crystallisation development, and bulk particle manipulation. Solid form evaluations are carried out before crystallisation and particle manipulation, which ultimately defines all final API attributes. These elements are summarised in the diagram below.
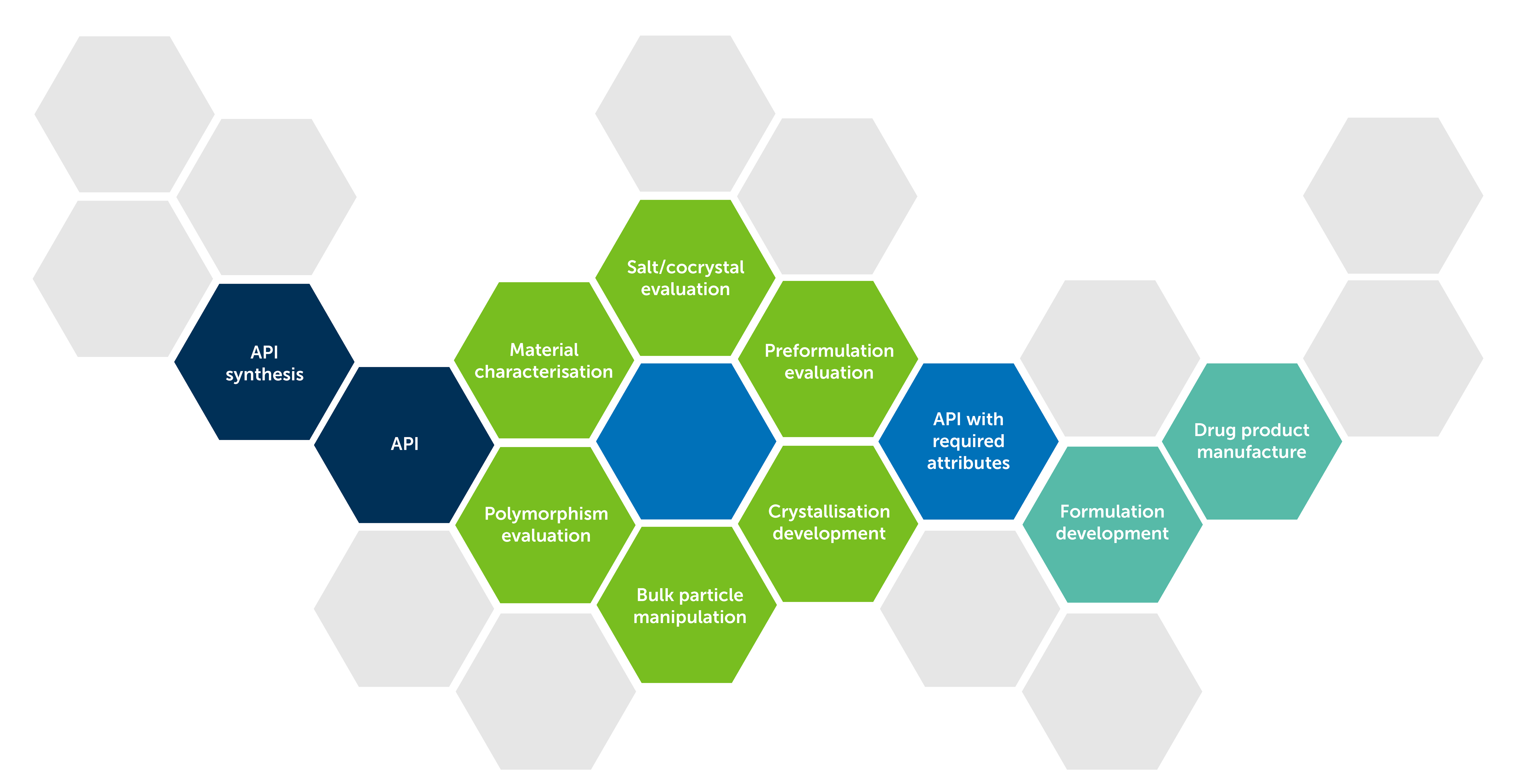
Let’s take a closer look at how each of these elements goes into identifying and progressing the most ideal solid form version:
When an API demonstrates appropriate characteristics, a salt or cocrystal screen can be considered to alter the properties of an API without changing its pharmacologically active moiety. Solubility, dissolution profile, and physicochemical characteristics like melting point and particle size can all potentially be improved by generating a salt or cocrystal version. For example, where crystallisation of the free API alone may fail to sufficiently improve chemical purity, implementing a salt or cocrystal formation could provide an opportunity for impurity control.
In addition, a salt screen isn’t just useful for early phase development or impurity control. A broad salt screen that evaluates a significant range of counter ions or co-formers and solvent mixtures can be utilised to generate intellectual property for API versions.
Whether investigating a salt version or the free API, polymorphism investigations are crucial for understanding the API’s propensity to polymorphism, as required by regulatory agencies.
Properties that can be improved by isolating a specific polymorphic form include efficacy, chemical and form stability, and impurity control.
Furthermore, polymorphism investigations allow scientists to understand the relationships between different forms and establish a form hierarchy to aid in identifying the ideal form for further development and manufacture. An expanded investigation that utilises a broad range of solvent and solvent mixtures can also be used to generate intellectual property.
Polymorphism investigations can be extensive; the more time scientists spend looking for different polymorphs, the more they are likely to uncover,so these investigations require a careful balance between costs, project timelines, and the risk of progressing an unsuitable form.
Since salt screens and polymorphism investigations may surface multiple potential candidates, additional assessments may be carried out to identify the most ideal form depending on the final product’s most critical attributes. These investigations may include:
- Solubility and stability in aqueous and biorelevant buffers
- Conditional storage under accelerated stability conditions
- Stability to compression and mechanical attrition
Pre-formulation evaluation is not always an independent step in solid form investigations; instead, some of it may be implemented during salt screens or polymorphism investigations where appropriate. If earlier screenings surface only one viable solid form candidate, pre-formulation evaluations may be skipped altogether.
The goal of effective crystallisation development is to generate the target API version of required chemical purity and particle characteristics with suitable recovery for drug product manufacture. It is used to control impurity levels, ensure the target API version is generated, and ensure a reproducible particle habit and size distribution.
These characteristics can have important impacts on unit operations like filtration, drying, particle size manipulation, and formulation, so controlled crystallisation that reproducibly affords the same quality material is crucial.
While an ideal crystallisation process would afford an API version with a suitable particle size distribution for manufacture, bulk particle manipulation may be employed to improve manufacturing or enhance final API performance. Bulk particle manipulation is a top-down operation, where techniques like milling and micronisation are applied to modify properties like shape, size distribution, and hygroscopicity.
Together, these five elements form the basis of a robust solid form development programme. But the most successful programmes also require extensive expertise, dedicated partnership, and comprehensive capabilities.
Solid state chemistry at Sterling
Solid form selection is a foundational component of API development, influencing everything from process efficiency, to solubility and stability, to regulatory success. By approaching solid form investigations and selection systematically, organisations can identify the best version early and mitigate potential challenges down the line.
At Sterling, our integrated approach to solid state chemistry allows us to help customers progress the ideal solid form for their drug substance. Our state-of-the-art Material Science Centre, located at our Cramlington, UK facility, is home to:
- Fully equipped solid form laboratory
- Milling and micronisation platforms with containment down to OEB4 class materials
- Instrumentation for XRPD, DSC, TGA, DVS, optical microscopy, hot stage microscopy, parallel synthesis, and more
- Experienced team of solid form development specialist
By coupling our extensive expertise with end-to-end API development and manufacturing capabilities, we collaborate with customers to reduce risk, increase manufacturing success, and develop high-quality APIs with confidence.
Ready to learn more about how we can support your project’s solid form requirements? Speak to a solid state expert today.




